

Антисинтетазний синдром: підходи до діагностики та лікування
Антисинтетазний синдром (АСС) — це відносно рідкісний симптомокомплекс невідомої етіології, який є найтяжчим підтипом поліміозиту / дерматоміозиту (ПМ/ДМ).
ПМ і ДМ — аутоімунні захворювання скелетних м’язів із переважним ураженням поперечно-посмугованих м’язів, із розвитком симетричної слабкості проксимальних м’язів і втягненням у патологічний процес периферичної нервової системи. Вони належать до системних захворювань сполучної тканини та об’єднуються спільним терміном — ідіопатична запальна міопатія (ІЗМ).
Клінічний перебіг АСС характеризуються гострим початком, наявністю ПМ/ДМ, інтерстиційним ураженням легень, синдромом Рейно, поліартралгією.
АСС асоціюється з наявністю специфічних імунологічних маркерів у сироватці крові — антисинтетазних антитіл (анти-Jo-1) до аміноацил транспортних РНК-синтетаз, функція яких полягає в каталізі процесів, пов’язаних із білковим синтезом.
Аміноацил-тРНК-синтетази транслюють білки з генетично транскрипованої мРНК. У 1999 році Wakasugi і Schimmel визначили, що ці синтетази можуть діяти як цитокіни, допомагаючи при загибелі клітин і утворенню запальних клітин. Подальші дослідження, підсумовані Ascherman у 2015 році, виявили вроджений і адаптивний імунітет у патогенезі антисинтетазного синдрому.
Дані статистики свідчать про ріст захворюваності на ДМ/ПМ у всьому світі, але особливо це фіксується в Південній Європі порівняно із Північною: відносна поширеність цих хвороб в Ірландії становить 0,08, а в Греції — 0,56.
У середньому частота розповсюдження ПМ/ ДМ в людській популяції становить від 2–10 випадків на 1 млн населення, у США — 5,5 на 1 млн населення.
Піки захворюваності припадають на дорослих осіб 50-літнього віку та дітей віком 10–15 років із співвідношенням жінок і чоловіків — 2–3:1.
Антисинтетазні антитіла
Виділяють такі міозит-специфічні антисинтетазні антитіла:
- Anti-Jo1
- Anti-PL7
- Anti-PL12
- Anti-OJ
- Anti-EJ
- Anti-KS
- Anti-Zo
- Anti-SC
- Anti-JS
- Anti-YRS
З антисинтетазних антитіл найчастіше ідентифікується anti-Jo-1. Менш поширені антисинтетазні антитіла — анти-треоніл (анти-PL7), анти-аланіл (анти-PL12), анти-ізолейцил (анти-ОJ) та анти-гільцил (анти-ЕJ).
Але відсутність антисинтетазних антитіл не виключає наявності АСС, бо рівні антитіл коливаються залежно від активності захворювання.
Клінічні симптоми та перебіг АСС відрізняються залежно від наявності антисентетазних антитіл.
Хамагучі та ін. провели дослідження 166 пацієнтів з наявністю антисинтетазних антитіл і продемонстрували, що у пацієнтів з анти-Jо-1, анти-ЕJ і анти-PL-7 найчастіше було клінічно діагностовано ДМ або ПМ, а у пацієнтів з анти-PL- 12 було виявлено аміопатичний ДМ або лише інтерстиціальне захворювання легень (ІЗЛ). Інтерстиціальне захворювання легень самостійно було поширене у пацієнтів з анти-KS та анти-OJ.
Дослідження у пацієнтів з анти-Jo-1 (n = 75) і анти-PL7 / PL12 (n = 20) у 2012 році виявило, що поширеність м’язових симптомів (слабкість та міалгія) була значно нижче у пацієнтів з анти-PL7 / PL12 порівняно з анти-Jo1.
Антисинтетазний синдром може проявлятися ізольовано з лихоманкою, артритом або ІЗЛ.
У багатьох публікаціях останніх років описано менш виражене м’язове ураження у хворих з антисинтетазним синдромом. Так, T. Fujisawa і співавтори вважають поганою прогностичною ознакою не тільки серопозитивність за антисинтетазними антитілами, а й низькі значення КФК (маркер м’язового некрозу), а незначне зниження м’язової сили в поєднанні з ІЗЛ розцінююється як фактор ризику швидкопрогресуючих ІЗЛ.
Z. Betteridge і співавтори спостерігали ураження легень за відсутності клінічно вираженої м’язової слабкості у хворих з іншими антисинтетазними антитілами: PL 12, KS, OJ.
Діагностичні критерії АСС
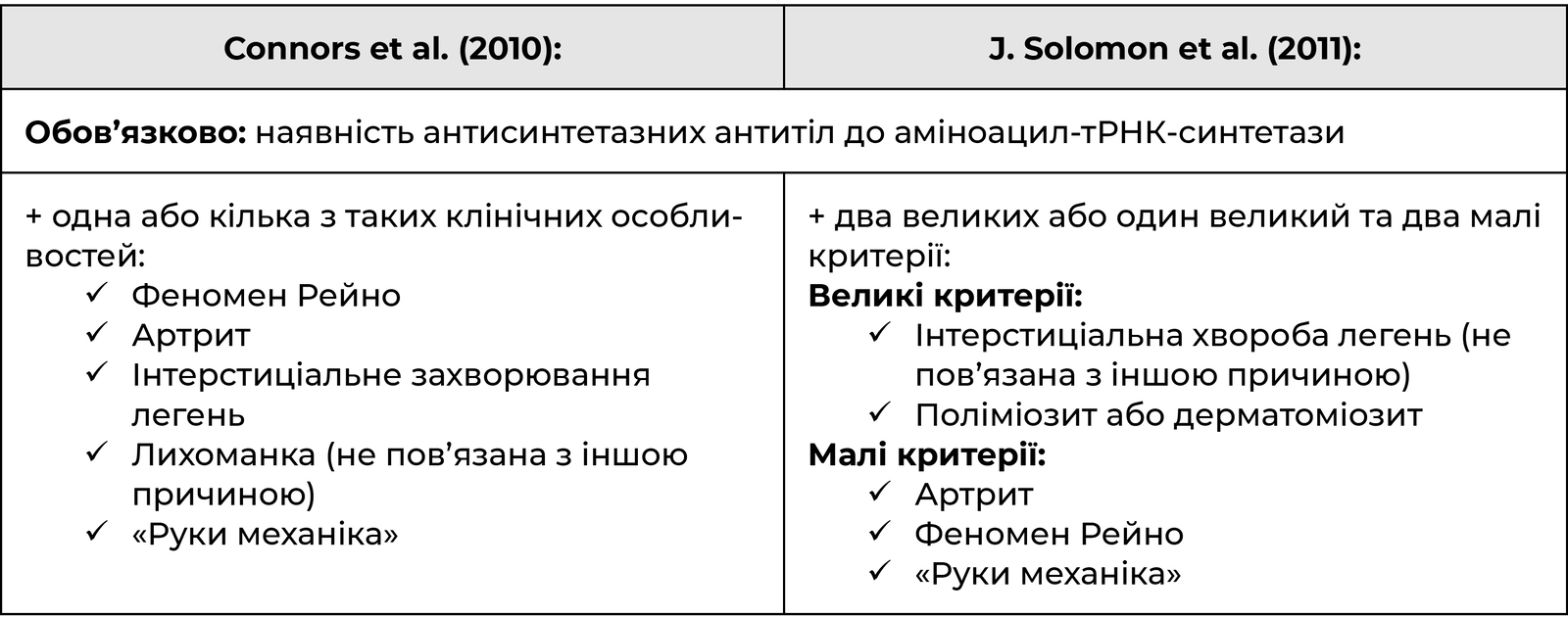
Основою патологічного процесу при ПМ/ДМ є запальна клітинна та периваскулярна інфільтрація скелетних м’язів Т-, В-лімфоцитами. У складі клітинного інфільтрату переважають CD8+-, CD4+-, CD28- лімфоцити, а також макрофаги, плазматичні, дендритні клітини.
Лаборатно-інструментальні показники м’язового пошкодження — рівень у сироватці крові креатинфосфокінази (КФК), альдолази, лактатдегідрогенази (ЛДГ) та зміни на електроміографії, які характерні для запальної міопатії. Підвищення рівня КФК може бути значно вираженим, у понад 50 разів порівняно з нормою.
Зазвичай рівні аспартатамінотрансферази (АсАТ) та аланінамінотрансферази (АлАТ) також підвищуються.
- Під час дослідження у 203 пацієнтів з антисинтетазним синдромом у 2013 році поширеність ІЗЛ становила 86 %, що частіше, ніж поширеність міозиту (73 %) або артралгії / артриту (60 %).
- Фактично, хворі на АСС можуть не мати класичних симптомів міопатії, як це спостерігається в разі захворювання на ПМ або міозит може з’явитись пізніше під час хвороби.
- Залежно від клінічних проявів, додаткове лабораторне обстеження може допомогти провести клінічну оцінку ступеня тяжкості та враження органів.
Інструментальні методи діагностики
- Рентгенографія ОГК.
- Комп’ютерна томографія з високою роздільною здатністю (використовується для моніторингу активності захворювання).
- Біопсія легень (рідко виконується у пацієнтів з АСС, бо діагноз, як правило, встановлюють шляхом синтезу результатів комп’ютерної томографії з високою роздільною здатністю, серологічних даних, об’єктивного обстеження та наявності симптомів захворювання).
- М’язову біопсію зазвичай не роблять, адже міозит часто відсутній або має субклінічний перебіг, а також немає даних про роль м’язової біопсії у діагностиці АСС.
Діагноз АСС встановлюють за допомогою міждисциплінарного підходу, синтезуючи ревматологічні ознаки захворювання та оцінюючи наявність захворювання легень, а також серологічних, рентгенографічних, а іноді і результатів біопсії легень або м’язів.
Пацієнти з антисинтетазним синдромом часто потребують мультимодальної імунодепресивної терапії для контролю м’язових та/або легеневих проявів захворювання.
Необхідна увага до побічних ефектів і ускладнень хронічної імуносупресивної терапії, а також пов’язаних із захворюваннями наслідків, серед яких можуть бути прогресуюча інтерстиціальна хвороба легень, що вимагає проведення трансплантації легень, та легенева гіпертензія.
Необхідно забезпечити ранню діагностику та відповідне лікування для покращення результатів у пацієнтів із АСС.
Лікування
- Основу лікування становлять ГК, які призначають в дозі не нижче 1 мг/кг/добу протягом 2,5–3 міс. із подальшим поступовим зниженням дози до підтримуючої.
- Проведені численні дослідження свідчать про позитивну динаміку та швидкість досягнення ремісії в разі раннього застосування імуносупресивних засобів, таких як: метотрексат (0,3 мг/кг/тиж), азатіоприн (2,5 мг/кг/добу), циклоспорин (3 мг/кг/добу), мофетилу мікофенолат. Введення циклофосфаміду (1 г/м2 поверхні тіла) щомісячно — препарат вибору при ІЗЛ.
- Раннє призначення терапії асоціюється зі сприятливішим прогнозом.
- За останні роки з’явилися повідомлення про успішне застосування біологічних препаратів, а саме: ритуксимабу при АСС, особливо — в разі резистентного до лікування перебігу.
- Неоднозначна роль анти-Jо-антитіл. З одного боку, їх підвищений титр свідчить про тяжкий перебіг, несприятливий прогноз захворювання, а з іншого — є предиктором адекватної відповіді на антиВ-клітинну терапію. Дані літератури свідчать про позитивний ефект і тривалу ремісію в разі застосування у таких хворих ритуксимабу.
Висновок
Таким чином, інтерстиціальне ураження легень займає центральне місце в клінічній картині антисинтетазного синдрому і визначає прогноз захворювання. Окремі дослідження дозволяють припустити, що виявлення анти-Jo 1-антитіл асоціюється, з одного боку, з наявністю важкого ІЗЛ, а з іншого — з відносно менш важким варіантом його перебігу. Не виключено, що різні види антисинтетазних антитіл можуть мати різне прогностичне значення.





